
Colloques
du Groupement des Anthropologistes de Langue Française (GALF)
Aquaron, R., Badens,
C., Bergé-Lefranc, J-L., Mansion, P., Mattei, J-F., Philip, N., Zarrouck, K.,
Naamane, A., Collignon, P., Burle, C., Rollier, B., Denis, D., 2004,
L’Albinisme oculocutané au Maghreb, à propos de 15 observations en Algérie,
Maroc et Tunisie. Antropo, 7, 55-61. www.didac.ehu.es/antropo
L’Albinisme
oculocutané au Maghreb, à propos de 15 observations en Algérie, Maroc et
Tunisie
Oculocutaneous
albinism in Maghreb, about 15 cases in Algeria, Morocco and Tunisia
R.
Aquaron1, C. Badens2, J-L. Bergé-Lefranc3, P.
Mansion1, J-F. Mattei4, N. Philip4, K.
Zarrouck5, A. Naamane5, P. Collignon6, C.
Burle6, B.Rollier7, D. Denis8
1Laboratoire
de Biochimie et Biologie Moléculaire, Faculté de Médecine, Marseille.
2Centre
d’Enseignement et de Recherche en Génétique Médicale, Faculté de Médecine ,
Marseille.
3Laboratoire
de Biologie Moléculaire, Hôpital de la Conception ,Marseille.
4Service
de Génétique Médicale, Hôpital d’enfants de la Timone , Marseille.
5Laboratoire
de Biochimie, Faculté de Médecine, Casablanca, Maroc.
6Services
de Génétique Médicale et de Néonatologie , Hôpital Font Pré, Toulon.
7Dermatologue
, Casablanca, Maroc.
8Service
d’ophtalmologie ,Hôpital Nord, Marseille.
Auteur chargé de la correspondance: Aquaron Robert , laboratoire de Biochimie et Biologie Moléculaire, Faculté de Médecine , 27 Boulevard jean Moulin , 13385 Marseille , Cedex 5 , France.
E-mail : robert.aquaron@medecine.univ-mrs.fr
Mots-clés: Albinisme oculocutané, tyrosinase, gène P, Algérie, Maroc, Tunisie
Keys-words: Oculocutaneous albinism , tyrosinase, P gene , Algeria , Morocco , Tunisia
Résumé
Nous rapportons l’étude clinique , biologique et moléculaire de 15 sujets atteints d’Albinisme OculoCutané (AOC) appartenant à 14 familles originaires de Tunisie (2 cas), d’Algérie (5 cas) et du Maroc (8 cas) dont 2 proviennent de couples mixtes : Maroc/Algérie et Maroc/France. La consanguinité est présente dans 11 familles et elle a pu être précisée dans 10 cas par le coefficient de consanguinité F= 1/16,1/32 et 1/64 . La recherche des mutations du gène de la tyrosinase caractéristique de l’AOCIA ou IB a pu être réalisée chez 9 sujets. .Dans 3 cas seulement des mutations faux sens ont été identifiées : 1 mutation homozygote (Y327C) chez un garçon marocain de phénotype AOCIA, 2 mutations hétérozygotes chez un garçon franco-marocain de phénotype AOCIB (V275F/R403S) et chez un garçon algérien de phénotype AOCIA (H404P/ ?).
Abstract
Clinical, biological and molecular characteristics of 15 patients with OculoCutaneous Albinism (OCA) from 14 families originated from Tunisia (2 cases), Algeria (5 cases) and Morocco (8 cases) were reported, 2 of them being from mixed couples : Morocco/Algeria and Morocco/France. Consanguineous matings occur in 11 families and coefficient of inbreeding was identified in 10 cases. F= 1/16,1/32 and 1/64 Identification of mutations of the tyrosinase gene characteristic of OCAIA or IB was performed in 9 subjects. Only in 3 cases, missense mutations were identified: a homozygous mutation (Y327C) in a moroccan boy with OCAIA phenotype; two heterozygous mutations in a franco-moroccan boy with OCAIB phenotype (V275/R403S) and in an algerian boy with OCAIA phenotype (H404P/ ?).
L’albinisme oculocutané (AOC) est une affection autosomique récessive qui se caractérise par une absence ou une diminution de la quantité de mélanine dans la peau , les cheveux , les poils et les yeux (iris, choroïde et épithélium pigmentaire de la rétine). L’absence ou la diminution de la biosynthèse de la mélanine dans l’oeil au cours du développement est associée à des modifications spécifiques (hypoplasie de la fovea , hypopigmentation du fond d’œil, anomalies de la réfraction et de routage des fibres optiques de la rétine au cortex optique) qui se manifestent dès la naissance par une acuité visuelle très faible , un nystagmus , une photophobie et quelquefois un strabisme. L’absence ou la diminution de la quantité de mélanine dans la peau est associée à une sensibilité accrue aux rayons UVA et UVB (érythème puis kératose actinique) et à une prédisposition aux cancers de la peau (carcinomes baso- et spino-cellulaires) principalement chez les sujets albinos africains de peau noire (Aquaron,2000; King et al., 2001).
Le caractère génétique de cette affection a été rattaché dès 1908 par Garrod à un déficit d’activité enzymatique. Il écrit "un enzyme intracellulaire est probablement absent chez les sujets albinos" et il mentionne que "la tyrosinase est responsable de la formation de mélanine" mais il ne fait pas de relation claire entre absence de tyrosinase et albinisme. Il a fallu attendre 1958 pour que Fitzpatrick et al. montrent , en utilisant la dopa réaction de Bloch que les follicules pileux de l’homme albinos contiennent des mélanocytes sans activité tyrosinasique. La fréquence générale de l’AOC est estimée à environ 1/20.000.
L’albinisme oculocutané a été divisé en 2 types en 1970 par Witkop et al., sur des critères cliniques (coloration de la peau , des cheveux et de l’iris) et biologiques en particulier l’incubation de bulbes de cheveux en présence de tyrosine et de dopa pour mettre en évidence la présence ou l’absence d’activité tyrosinase :coloration brune pour le type tyrosinase-positif (tyr-pos) , absence de coloration pour le type tyrosinase-négatif (tyr –neg). Cette classification phénotypique a été très utile pour distinguer ces deux types d’AOC mais n’a pas permis d’élucider le caractère génétique de chacun de ces deux types. Il a fallu attendre l’identification, le clonage et l’étude des mutations du gène de la tyrosinase et du gène P pour proposer une classification moléculaire.
Le type tyr -pos est actuellement appelé AOC2 (OMIM : 203200).Il correspond à une activité tyrosinasique in vitro mais une absence d’activité in vivo vraisemblablement due à un pH défavorable. Il se caractérise cliniquement par la présence d’une peau blanche ou légèrement pâle qui a la possibilité de bronzer, de cheveux blonds et d’iris bleu ou plus pigmenté (noisette, vert). Il est dû à des mutations du gène P.
Le type tyr-neg est dénommé AOC1 (OMIM: 203100) et résulte de mutations du gène de la tyrosinase . Sa fréquence générale est d’environ 1/40.000 et on le trouve principalement chez les sujets caucasiens. Ce type est lui-même subdivisé en 2 sous-groupes: l’AOC1A qui se caractérise par une activité tyrosinase nulle d’où une absence totale de mélanine (ancien tyr-neg) et cliniquement par une peau blanche, des cheveux blancs et un iris bleu pendant toute la vie .L’exposition au soleil produit un érythème mais jamais de bronzage. L’AOC1B dans lequel existe une activité tyrosinase résiduelle (5-10% de l’activité normale) est caractérisé à la naissance par des cheveux blancs mais qui développent dans la 1ère décade de la vie une petite quantité de pigments (cheveux blonds, iris bleu ou noisette). L’exposition au soleil produit un léger bronzage.
|
Cas |
Nom |
Sexe |
A.D |
Age |
Pays |
Alb/Tot |
F* |
Couleur Iris |
Couleur Cheveux |
Tyr** |
Mutations gène Tyr |
|
1 |
Bou… |
M |
1978 |
4 |
Algérie |
1/3 |
1/32 |
Bleu |
Blond |
Neg |
NR |
|
2 |
Ket… |
F |
1984 |
10 |
Algérie |
1/3 |
1/64 |
Bleu |
Blond |
Neg |
NR |
|
3 |
Ham… |
M |
1991 |
1 |
Algérie |
1/2 |
? |
Bleu |
Blanc |
Neg |
H404P/R402Q |
|
4 |
Skl… |
F |
1995 |
5 |
Algérie |
1/2 |
1/64 |
Gris-bleu |
Blond |
Neg |
Non trouvée |
|
5 |
Amr… |
F |
1993 |
0.5 |
Maroc |
1/6 |
1/32 |
Bleu |
Blond |
Pos |
NR |
|
6 |
Lag… |
M |
1998 |
3 |
Maroc |
1/2 |
1/64 |
Gris-bleu |
Blanc |
Neg |
Y327C/Y327C |
|
7 |
Ane…L |
F |
1998 |
1 |
Maroc |
2/3 |
1/32 |
Noisette |
Blanc |
Neg |
Non trouvée |
|
8 |
Ane…S |
F |
1999 |
1 |
Maroc |
2/3 |
1/32 |
Noisette |
Blanc |
Neg |
Non trouvée |
|
9 |
Err… |
M |
1997 |
32 |
Maroc |
1/7 |
1/16 |
Noisette |
Blond |
Pos |
Non trouvée |
|
10 |
Ram… |
M |
1997 |
18 |
Maroc |
1/4 |
1/32 |
Gris-bleu |
Blond |
NR |
NR |
|
11 |
Ela… |
F |
1995 |
15 |
Maroc |
3/3 |
0 |
Gris-bleu |
Blond |
Pos |
NR |
|
12 |
Rmi… |
M |
1998 |
2 |
Mar/Alg |
1/2 |
0 |
Noisette |
Blond |
Neg |
Non trouvée |
|
13 |
Hou… |
M |
1991 |
1 |
Mar/Fra |
1/1 |
0 |
Bleu |
Blond |
Neg |
V275F/R403S |
|
14 |
Lak… |
F |
1983 |
11 |
Tunisie |
2/11 |
1/16 |
Gris-bleu |
Blanc |
NR |
NR |
|
15 |
Bou… |
M |
1995 |
21 |
Tunisie |
1/3 |
0 |
Gris-bleu |
Blond |
Pos |
Non trouvée |
Tableau 1. Caractéristiques cliniques, biologiques et moléculaires de 15 sujets albinos originaires d'Algérie, du Maroc et de Tunisie. A.D = Année au moment du Diagnostic; F*= coefficient de consanguinité, Tyr **présence (tyr-pos) ou absence (tyr-neg) d'activité tyrosinasique dans les bulbes de cheveux; Alb/Tot= rapport entre le nombre de sujets albinos et le nombre total d'enfants; NR= Non réalisé.
Table 1. Clinical, biological and molecular characteristics form 15 albinos patients issued from Algeria, Morocco and Tunisia. A.D = Year at Diagnosis; F*= inbreeding coefficient; Tyr**= Presence (tyr-pos) or absence (tyr-neg) of tyrosinase activity in hair bulb test; Alb/Tot= ratio between albinos and total number of children; NR= Not realized.
L’AOC a été décrit
dans pratiquement tous les pays du monde (King et al., 2001) mais curieusement
les observations relatives au Maghreb sont rares. Les 2 observations décrites
dans la littérature rapportent la présence d’épithéliomas baso- et
spino-céllulaires chez 2 sujets algériens albinos âgés respectivement de 25 et
64 ans (Hadida et al., 1966; Esteve, 1981). En raison de la présence importante
de sujets maghrébins dans la région Provence- Alpes- Cote d’Azur., nous avons
entrepris un recensement de ces sujets albinos.
Notre travail a
consisté en l’étude clinique, biologique et moléculaire de 15 sujets albinos
appartenant à 14 familles originaires de Tunisie (2), d’Algérie (5) et du Maroc
(8) avec 2 couples mixtes: Maroc/Algérie et Maroc/France. Les principales
caractéristiques démographiques, cliniques, biologiques et moléculaires sont
brièvement résumées dans le tableau 1. Les observations de certains de ces
sujets ont été décrits avec plus de détail par Naamane en 2000 et par Mansion
–Mixayphonh en 2003.
Les patients 1, 2, 3, 4, 7, 8, 13 et 14 ont été examinés à Marseille, les sujets 5 et 12 à Toulon, les patients 6, 9, 11, 15 à Casablanca et 10 à Marrakech.
On a observé 7 filles et 8 garçons âgés de 6 mois à 32 ans; cette répartition voisine de 50% correspond bien à une affection non liée au sexe.
Dans 9 familles il existe une consanguinité connue: F=1/16 dans 2 familles (union entre cousins germains), F=1/32 dans 4 familles (union entre demi-cousins germains ou cousins germains moins un: le père est cousin germain avec la mère de sa femme), F=1/64 dans 3 familles (union entre cousins issus de germains). Comme dans toute affection de transmission autosomique récessive, l’AOC est plus fréquent dans les familles consanguines. Le taux de consanguinité au Maghreb est évalué à: 29% au Maroc, 40% en Algérie et 48% en Tunisie (Hoodfar et Teebi,1996).
Le nombre de sujets albinos par rapport aux sujets normaux est de 18/52 soit une fréquence de 1/3 (34%) supérieure à la fréquence théorique de 1/4 (25%) en raison du biais de recrutement des familles par la présence d’un sujet albinos.
L’activité
tyrosinasique dans les bulbes de cheveux a été pratiquée 13 fois (9 tyr-neg et
4 tyr-pos) selon la technique décrite par Nixon en 1973: incubation de cheveux
de type anagène avec le bulbe intact dans une solution contenant de la
L.tyrosine 2,5mM et de la L.Dopa 0,1mM dans un tampon phosphate 0,1M de pH 6,8
pendant 5 heures à l’obscurité.
La recherche des
mutations du gène de la tyrosinase a été réalisée après amplification de l’ADN
génomique par PCR, en observant une anomalie de migration après électrophorèse
(SSCP: single strand conformational polymorphism) et/ou séquencage des 5 exons
de la tyrosinase. Sur 9 échantillons analysés aucune mutation n’a été mise en
évidence dans 6 cas. Chez 3 sujets on a trouvé des mutations faux
sens c.a.d le remplacement d’un acide aminé par un autre. Le taux de
réussite est seulement de 3/9 soit 33%, taux plus faible que dans des études
portant sur un nombre plus élevé de sujets albinos: 19/45 soit 42% en France et
au Canada (Camand et al., 2001); 32/74 soit 43% en Allemagne (Passmore et al.,
1999) et 102/120 soit 85% aux USA (King et al., 2003). Les mutations non
trouvées responsables de l’AOC peuvent se situer dans une zone du gène non
explorée ou dans un autre gène participant également à la biosynthèse des
mélanines. Les 3 sujets présentent: une mutation à l’état homozygote (Y327C)
chez un garçon marocain, N°6 et Figure 2, deux mutations à l’état de double
hétérozygote: chez un garçon franco-marocain, N° 13 et Figure 3 (V275F/R403S)
et chez un garçon algérien N°3 (H404P / ?). La mutation Y327C qui
transforme un résidu de tyrosine (Y) par un résidu de cystéine (C) est une
mutation nouvelle. Les mutations V275F et R403S ont déjà été décrites
respectivement par Giebel et al. en 1991 et par Tripathi et al. en 1992. La mutation
V275F, Valine (V) remplacée par Phenylalanine (F) située sur l’exon 2 était
présente chez la mère d’origine française. La mutation R403S, Arginine (R)
remplacée par Sérine (S) située sur l’exon 4 était présente chez le père
d’origine marocaine. Ces 2 mutations sont caractéristiques de l’AOCIB. La
mutation H404P, Histidine (H) remplacée par Proline (P) située sur l’exon 4 a
déjà été décrite par Oetting et al. en 1998. Ce sujet présentait également un
polymorphisme R402Q c.a.d qu’en position 402 on peut trouver soit l’Arginine
(R) dans 85% des cas, soit la glutamine (Q) dans 15% des sujets. Ce
polymorphisme décrit par Tripathi et al. en 1991 est associé avec une
activité tyrosinasique normale à 31° et une activité très réduite à 37°. Il est
intéressant de noter que le polymorphisme (R402Q) et les 2 mutations (R403S et
H404P) se suivent dans la séquence et se situent à proximité du site B du
cuivre indispensable à l’activité de la tyrosinase qui transforme la tyrosine
en dopa puis en dopaquinone. Les relations phénotype-génotype ont été bien
étudiées par Passmore et al. 1999 et par King et al. 2003. Pour le
premier une corrélation phénotype AOCI et mutation du gène de la tyrosinase est
trouvée dans 25 cas mais 10 sujets du phénotype AOC2 présentent des mutations
sur le gène de la tyrosinase. Inversement 3 sujets de phénotype OCAI possèdent
des mutations du gène P. Pour King et al. 2003, qui se sont surtout
focalisés sur la distinction OCAIA-OCAIB, le critère phénotypique le plus
important pour diagnostiquer l’OCAIA est la présence à la naissance et durant
toute la vie de cheveux blancs ce qui rend compte des 85% de réussite dans
l’identification de mutations du gène de la tyrosinase. La jeune fille tunisienne
(Figure 4 , N°14) qui présente des cheveux blancs à l’âge de 10 ans est
certainement OCAIA mais ni ses bulbes de cheveux ni le gène de la tyrosinase
n’ont pu être testées. La jeune fille algérienne (Figure 1 , N° 2) qui
possédait à la naissance des cheveux blancs puis blonds à l’âge de 10 ans ainsi
qu’un test bulbaire négatif est vraisemblablement OCAIB mais le génotype n’a
pas pu être contrôlé.
La recherche des
mutations du gène P s’est limitée à la mise en évidence de la délétion 2,7kb
dans 2 cas où le phénotype clinique pouvait être soit OCAIB soit OCA2 et elle
était négative.
L’intérêt de mettre en évidence les mutations des gènes P et de la tyrosinase des sujets albinos est de permettre dans une famille la caractérisation des sujets normaux hétérozygotes (parents, frères et sœurs du propositus) et en cas de grossesse ultérieure de réaliser un diagnostic prénatal (Shimizu et al., 1994; Rosenmann et al., 1999).
|
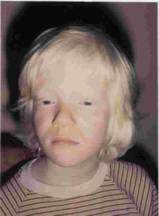
Figure 1. Cas N° 2: fille algérienne albinos âgée de 10 ans de phénotype clinique AOC1B ou AOC2 (cheveux blonds et iris bleu), Marseille,1984. Figure 1. Case N° 2: algerian albino girl 10 years old with a clinical phenotype OCA1B or OCA2 (blond hair and blue iris) Marseille, 1984 |
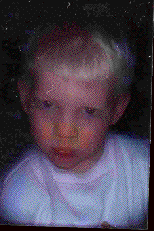
Figure 2. Cas N° 6: garçon albinos marocain âgé de 4 ans de phénotype clinique (cheveux blancs et iris bleu) et moléculaire (mutation homozygote du gène de la tyrosinase Y327C/Y327C) caractéristique de l’AOC1A, Casablanca, Maroc, 1999. Figure 2. Case N°6: moroccan albino boy 4 years old with a clinical (white hair and blue iris) and molecular (homozygous mutation of the tyrosinase gene Y327C/Y327C) characteristic of OCA1A, Casablanca, Morocco, 1999.
|
|
Figure 3.
Cas N° 13: garçon albinos franco-marocain âgé de 2 ans de phénotype clinique
(cheveux blonds et iris bleu) et moléculaire (double mutation hétérozygote du
gène de la tyrosinase V275F/R403S) de type AOC1B, Marseille, 1993. Figure 3. Case N° 13: franco-moroccan albino boy 2 years old with a clinical (blond hair and blue iris) and molecular (double heterozygous mutation of the tyrosinase gene V275F/R403S) of type OCA1B, Marseille, 1993. |
Figure
4. Cas N° 14: fille albinos tunisienne âgée de 10 ans de
phénotype clinique AOC1A (cheveux blancs et iris gris-bleu), Marseille 1983. Figure 4. Case N° 14: tunisian albino girl 10 years old with a clinical phenotype OCA1A (white hair and grey-blue iris), Marseille, 1983. |


On peut trouver la liste des mutations du gène de la tyrosinase sur la banque de données: http://www.cbc.umn.edu/tad/index.html
Remerciements. Les auteurs ont vivement apprécié la collaboration de tous les sujets albinos et leurs familles et celle de Mr. Yves Mourayre pour son assistance technique. Ils remercient l’équipe du Pr. R. King, W. Oetting et J. Fryer, Department of Medicine; University of Minnesota, Minneapolis, Minn., USA pour le chaleureux accueil réservé à l’un d’entre nous (R.A) pendant un court séjour à Minneapolis et pour l’identification des mutations du gène de la tyrosinase: V275F, R403S et H404P.
Références
Aquaron, R., 2000, L’albinisme humain: aspects cliniques, génétiques, cellulaires, biochimiques et moléculaires. Médecine Tropicale, 60, 331-341.
Camand, O., Marchant, D., Boutboul, S., Péquignot, M., Odent, S., Dollfus, H., Sutherland, J., Levin, A., Menasche, M., Marsac, C., Dufier, J-L., Heon, E., et Abitbol, M., 2001, Mutation analysis of the tyrosinase gene in oculocutaneous albinism. Mutations in brief, N°409, On line, Human Mutations, 17, 352-357.
Esteve, E., 1981, Albinismes oculo-cutanés,épithéliomas cutanés. Discussion à propos d’un cas. Thèse de Doctorat en Médecine, Toulouse,°73.
Fitzpatrick, T. B., Brunet, P., et Kukita, A., 1958, The nature of hair pigment. Dans The biology of hair growth , édité par W. Montagna et R. A.Ellis (New-York, Academic Press) p. 255-303.
Garrod, A., 1908, The croonian lectures on inborn errors of metabolism. General and introductory. Lancet, ii, 1-7.
Giebel, L.B., Tripathi, R.K., Strunk, K.M., Hanifin, J.M., Jackson, C.E., King, R.A., et Spritz, R.A., 1991, Tyrosinase gene mutation associated with type IB ("Yellow") oculocutaneous albinism. American Journal of Human Genetics, 48, 1159-1167.
Hadida, E., Sayag, J. et Tasso, M.F., 1966, Albinos de 25 ans. Peau rhomboïdale. Lésions kératosiques, dégénérescence épithéliomateuse. Bulletin de la Société Française de Dermatologie et de Syphiligraphie, 73, 294-295.
Hoodfar, E., et Teebi, A.S., 1996, Genetic referrals of middle eastern origin in a western city : inbreeding and disease profile. Journal of Medical Genetics, 33, 212-215.
King, R.A., Hearing, V.J., Creel, D.J., et Oetting, W.S., 2001, Albinism. Dans The metabolic basis of inherited disease, 8ème édition, edité par C.R. Scriver, A.L. Beaudet, W.S. Sly et D. Valle (New York, McGraw-Hill) p. 5587.
King, R.A., Pietsch, J., Fryer, J.P., Savage, S., Brott, M.J., Russell-Eggitt, I., Summers, C.G., et Oetting, W.S., 2003, Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Human Genetics, 113, 502-513.
Mansion-Mixayphonh, P., 2003, L’albinisme oculocutané dans la région marseillaise: à propos de 26 observations. Thèse de doctorat en Médecine, Marseille, pp. 212.
Naamane, A., 2000, L’albinisme oculocutané. Au Maroc: à propos de 7 cas. Thèse de doctorat en Médecine, Casablanca, pp. 104.
Nixon, P.F., 1973, The "red-skinned" New Guinean: distinctive melanocytes. Dans Mechanism in pigmentation, édité par V.J. McGovern et P. Russell (Basel, Karger) vol. 1, p. 6-13.
Oetting, W.S., Fryer, J.P., et King, R.A., 1998, Mutations of the tyrosinase gene associated with tyrosinase related oculocutaneous albinism (OCAI). Mutations in brief N° 204 On line. Human Mutations, 12, 433-434.
Passmore, L.A., Kaesmann-Kellner, B., et Weber, B.H., 1999, Novel and recurrent mutations in the tyrosinase gene and the P gene in the german albino population. Human Genetics, 105, 200-210.
Rosenmann, R.E., Rosenmann, A., Ne’eman, Z., Lewin, A., Bejarano-Achache, I. , et Blumenfeld, A., 1999, Prenatal diagnosis of oculocutaneous albinism type I: review and personal experience. Pediatric Developmental Pathology, 2, 404-414.
Shimizu, H., Niizeki, H., Suzumori, K., Aozaki, R., Kawaguchi, R., Hikijii, K., et Nishikawa, T., 1994, Prenatal diagnosis of oculocutaneous albinism by analysis of the fetal tyrosinase gene. Journal of Investigative Dermatology, 103, 104-106.
Tripathi, R.K., Giebel, L.B., Strunk, K.M., et Spritz, R.A., 1991, A polymorphism of the human tyrosinase gene is associated with temperature-sensitive enzyme activity. Gene Expression, 1, 103-110.
Tripathi, R.K., Strunk, K.M., Giebel, L.B., Weleber, R.G., et Spritz, R.A., 1992, Tyrosinase gene mutations in type I (tyrosinase-deficient) oculocutaneous albinism define two clusters of missense substitutions. American Journal of Medical Genetics, 43, 865-871.
Witkop, C., Nance, W., Rawls, R., et White, J., 1970, Autosomal recessive oculocutaneous albinism in man: evidence for genetic heterogeneity. American Journal of Human Genetics, 22, 55-74.