Valero Almingol, A., Peña, J.A., 2021 Origen de la variación clinal en el genoma humano europeo. Antropo, 46, 41-56. www.didac.ehu.es/antropo
Origen de la variación clinal en el genoma humano europeo
Origin of clinal variation in the European human genome
Alba Valero Almingol, Jose A. Peña
Dpto. Genética, Antropología Física y Fisiología Animal, Fac. Ciencia y Tecnología, Universidad del País Vasco, UPV/EHU. avalero015@ikasle.ehu.eus, joseangel.pena@ehu.eus
Palabras clave: Clinas, Europa, GWCline
Keywords: Clines, Europe, GWCline
Resumen
En este trabajo se ha empleado una base de datos de 621.799 SNPs analizados en individuos de varias poblaciones, con el fin de caracterizar las clinas presentes en el genoma humano europeo. Muchas clinas se producen por la mezcla de dos poblaciones de diferente origen, por lo que resultan herramientas apropiadas para comprobar la existencia de grandes oleadas migratorias en un continente. Se ha tratado de determinar el posible origen en el tiempo de las tendencias observadas, adscribiéndolas a las tres principales oleadas de migración constatadas en la historia del continente: la recolonización postglacial, la expansión neolítica de los granjeros de Anatolia y la expansión de la cultura Yamnaya desde la estepa póntica.
Tras excluir las clinas que han sido potencialmente producto de la selección natural, el resultado ha sido una mayoría de clinas con orientación latitudinal, probablemente resultante de la difusión de las tres oleadas migratorias mencionadas. Además, en el cromosoma Y se han detectado varias clinas longitudinales, probablemente correspondientes al haplogrupo R1b-M269, que a pesar de su orientación parece haberse expandido por Europa junto con la cultura Yamnaya.
Abstract
In this work we have used a database of 621,799 SNPs analyzed in individuals from various populations, in order to characterize the clines present in the European human genome. As many clines are created because of the mixture of two populations of different origin, they are important tools to detect the presence of major migration waves in a continent. To try and determine the origin in time of the observed tendencies, these have been ascribed to the three main historical migrations of the continent: the postglacial recolonisation, the expansion of Anatolian farmers during the Neolithic, and the expansion of the Yamnaya culture from the Pontic steppe.
After excluding clines that have potentially been the product of natural selection, the result has been a majority of latitudinally oriented clines, probably as a result of the spread of the three migratory waves mentioned above. In addition, several longitudinal clines have been detected on the Y chromosome, probably corresponding to haplogroup R1b-M269, which despite its orientation seems to have spread throughout Europe together with the Yamnaya culture.
Introducción
La variación genética humana se lleva estudiando más de un siglo mediante marcadores indirectos como proteínas y, más recientemente, mediante ADN (Cavalli-Sforza, 2005). El conocimiento de la genética de las poblaciones es de enorme importancia, ya que resulta útil a diferentes niveles como el estudio de la historia humana, el estudio de enfermedades con origen genético o incluso el desarrollo de fármacos específicos.
A partir de estos estudios, se ha visto que la variación genética en humanos está enormemente relacionada con la geografía. Las clinas, entendidas como la variación lineal gradual de un carácter a lo largo de la geografía, se crean principalmente por dos motivos. Por un lado, la selección natural suele ser responsable de crear clinas en genes concretos; por otro lado, las expansiones démicas generan clinas para varios genes (Cavalli-Sforza, 2005), a causa de la mezcla de dos poblaciones de distinto origen. De esta forma, podrían ser una fuente para explicar la variación existente en las poblaciones humanas. De hecho, la información genética se ha usado para tratar de reconstruir la historia humana (Lawson Handley et al., 2007; Cavalli-Sforza, 2005), por lo que el estudio de genomas completos de diferentes poblaciones podría arrojar algo de luz sobre las oleadas migratorias ocurridas en los diferentes continentes.
Las migraciones caracterizan la historia evolutiva de los humanos modernos y vienen motivadas por diferentes razones como las innovaciones culturales o los cambios ambientales (Pinhasi et al., 2012). Sin embargo, es importante entender las migraciones no como eventos bruscos y direccionales, sino que en muchos casos son procesos que se extienden a lo largo de un gran periodo de tiempo y no tienen por qué ser rápidos (Skoglund y Mathieson, 2018). En Europa, la estructura genética actual forma un continuo a lo largo del continente, y su historia se caracteriza por distintas oleadas migratorias. El patrimonio genético europeo actual proviene de tres poblaciones distintas. En primer lugar, cazadores-recolectores del oeste de Europa, procedentes de África, que se dispersaron en grupos por el continente. En segundo lugar, la expansión de los agricultores de Anatolia durante el Neolítico. En tercer lugar, individuos procedentes de la estepa póntica que se expandieron por Europa durante la Edad del Bronce, asociados a la cultura Yamnaya, que a su vez poseían ancestría principalmente de cazadores recolectores del este de Europa y cazadores recolectores del Cáucaso (Skoglund y Mathieson, 2018), pero también una parte de Próximo Oriente, similar a la composición actual de europeos del este y del norte de Europa (Saag et al., 2021). Con todo ello, las frecuencias alélicas actuales en Europa muestran una gran correlación con la geografía (Lazaridis et al., 2014; Gokcumen y Frachetti, 2020).
La colonización de Europa por los Humanos Anatómicamente modernos ocurrió hace unos 42.000-45.000 años (Pinhasi et al., 2012; Skoglund y Mathieson, 2018). Posteriormente, hace entre 25.000 y 19.000 años ocurrió el Último Máximo Glacial (Fu et al., 2016). Durante esta época el norte y centro de Europa quedaron cubiertos de hielo o permafrost, por lo que los recursos en esa área eran escasos y buena parte fue abandonada. Esto conllevó que las poblaciones humanas tuviesen que quedar relegadas a los refugios glaciales, que se encontraban en el sur de Europa: el refugio francocantábrico, ubicado entre el sudoeste de Francia y la Península Ibérica, el refugio de los Balcanes y el refugio del sur de Ucrania (Torroni et al., 1998; Jones et al., 2017). Estos lugares, por no estar cubiertos de hielo, poseían condiciones climáticas adecuadas para la existencia de animales y plantas (Pinhasi et al., 2012). Tras el fin del periodo glacial y el derretimiento de los hielos, se produjo una repoblación del centro y norte de Europa, que provino probablemente de los refugios de centroeurasia y el sur de Europa.
La segunda migración fue la de los agricultores de Anatolia durante el Neolítico. La agricultura surgió en Oriente Próximo entre el 10.000 y el 9.000 AC (Lazaridis et al., 2016), y constituyó un acontecimiento importante, ya que las poblaciones pudieron expandirse debido a la abundancia de comida que conllevó (Rosser et al., 2000). Además, vino acompañado de grandes cambios para el entorno y la organización social (Isern et al., 2017). Los primeros agricultores se expandieron por Europa hace unos 8.500 años, primero al sur y luego al norte (Gokcumen y Frachetti, 2020; Skoglund y Mathieson, 2018). Esta migración fue progresiva, alcanzando extremos de Europa como Iberia hace 7.300 años y Escandinavia hace unos 4.900 años (Skoglund y Mathieson, 2018). Durante décadas existieron dos hipótesis sobre cómo ocurrió la expansión por Europa. La hipótesis de difusión démica sugería que la expansión fue tanto genética como cultural, mientras que, según la hipótesis de difusión cultural, la expansión cultural habría ocurrido sin un gran movimiento poblacional, de lo que se deduciría que la diversidad genética en Europa provendría principalmente del paleolítico (Rosser et al., 2000). Sin embargo, se ha visto que la difusión démica fue más importante que la difusión cultural (Isern et al., 2017), de modo que la expansión condujo no solo a la difusión de ideas, sino que produjo un profundo cambio en el patrimonio genético (Jones et al., 2017). Por otro lado, los cazadores-recolectores no fueron completamente reemplazados. Tras la expansión por Europa ambas poblaciones se mezclaron durante varios milenios, y la mayoría de poblaciones europeas tendrían ancestría de ambas poblaciones, con aproximadamente un 10-25% de ancestría de cazadores-recolectores (Hofmanová et al., 2016; Skoglund y Mathieson, 2018).
La tercera migración fue la de los hombres de las estepas durante la Edad de Bronce, que comenzó hace unos 5.000 años (Skoglund y Mathieson, 2018). Se ha observado que durante este periodo las poblaciones europeas, que se encontraban compuestas por una mezcla de ancestría de cazadores-recolectores y agricultores neolíticos, recibieron un flujo genético de la zona del Cáucaso, lo que coincide con la expansión de la cultura Yamnaya desde la estepa póntica (Allentoft et al., 2015). Esta expansión probablemente supuso también la expansión de las lenguas Indoeuropeas, de forma que encajarían los datos lingüísticos, arqueológicos y genéticos (Allentoft et al., 2015). Además, fue una expansión muy rápida, propiciada por los carros y los caballos que portaban (Haak et al., 2015). Por otro lado, se ha observado que su contribución en los genomas masculinos fue muy importante (Saag et al., 2021). De hecho, habrían sustituido a todos los linajes masculinos previos de la Península Ibérica (Olalde et al., 2019).
Hoy en día, los tres componentes de ancestría se reparten de manera dispar a lo largo de Europa. Como se puede observar en la Figura 1, las poblaciones del norte de Europa son las que poseen un mayor componente Yamnaya, mientras que las poblaciones del sudeste de Europa poseen un mayor componente neolítico. Sin embargo, el componente de los cazadores-recolectores de Europa occidental se encuentra en menor proporción que ambos en la mayoría de poblaciones.
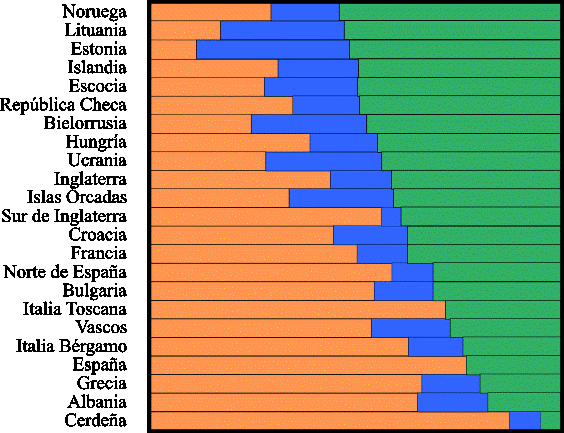
Figura 1. Ancestría de los principales grupos que componen el genoma humano europeo a lo largo del continente (Haak et al., 2015). En naranja, componente Neolítico, en azul componente cazador-recolector del oeste de Europa y en verde, componente Yamnaya.
Figure 1. Ancestry of the main groups that make up the European human genome across the continent (Haak et al., 2015). In orange, Neolithic component, in blue Western European hunter-gatherer component and in green, Yamnaya component.
El objetivo de este trabajo es detectar las clinas presentes en el genoma humano europeo, comprobar su orientación y deducir su origen. Se parte de la hipótesis de que si tanto la recolonización postglacial del continente como la migración neolítica ocurrieron esencialmente de sur a norte y la migración y la expansión de los Yamnaya ocurrió a grandes rasgos de norte a sur, la mayoría de las clinas deberían ser latitudinales, mientras que las clinas longitudinales deberían ser minoritarias.
Con el fin de lograr estos objetivos se empleará el programa GWCline (Peña, 2022) para la detección de las clinas y su orientación a partir de una base de datos de 621.799 SNPs y posteriormente se tratará de asignar cada tendencia observada a presiones selectivas o bien a las oleadas migratorias que conforman la ancestría de las poblaciones europeas actuales.
Materiales y métodos
Base de datos
La base de datos utilizada incluye individuos de diferentes poblaciones del mundo procedentes del Human Genome Diversity Project (Cavalli-Sforza, 2005) y se compone de los genotipos de 621.799 SNPs analizados por Lazaridis et al. (2016). Se eligieron individuos de distintas poblaciones europeas, procurando que se distribuyeran de manera homogénea por todo el continente.
Se seleccionaron un total de 298 individuos, sin atender a la proporción de sexos. Las poblaciones incluidas en el estudio fueron: Albania, Bielorrusia, Bulgaria, Cerdeña, Chipre, Croacia, Escocia, España, Finlandia, Francia, Georgia, Grecia, Hungría, Inglaterra, Islandia, Italia, Lituania, Malta, Noruega, País Vasco, República Checa, Rumanía, Rusia, Sicilia y Ucrania.
Programas
Para el tratamiento de datos se emplearon los programas GWCline (Peña, 2022) y GenoCline (Peña et al., 2022).
El programa GWCline se empleó para la detección de clinas a partir de una base de datos disponible en formato EIGENSOFT. Este formato se compone de tres tipos de ficheros. En primer lugar, los ficheros de individuos, que contienen la etiqueta correspondiente, el sexo y su origen. En segundo lugar, los ficheros de genomas, que contienen los genotipos individuales; en este fichero se codifica con un número el genotipo de cada individuo y SNP, existiendo cuatro posibilidades: el número 0 indica que no hay ninguna copia del alelo principal, el número 1 indica que existe una única copia del alelo principal, el número 2 indica que existen dos copias del alelo principal y el número 9 indica que no se ha podido hacer una lectura correcta. En tercer lugar, los ficheros de SNPs contienen el nombre del SNP, el cromosoma donde se encuentra, su posición dentro del cromosoma, el alelo principal y el alelo alternativo. Además, se introdujeron las coordenadas de cada individuo, estableciendo como referencia la capital del país o de la zona geográfica a la que pertenecían.
GWCline proyecta la posición de cada individuo sobre un eje de coordenadas que rota 360 grados, en iteraciones de 1 grado. En cada iteración, se calcula el coeficiente de correlación entre el vector de coordenadas y el vector de genotipos. Finalmente, si existe correlación significativa para una o varias orientaciones, se selecciona aquella que muestra una significación mayor. Una descripción más detallada del método puede encontrarse en Peña et al. (2022).
Puesto que se realizaron un gran número de pruebas, una parte de las clinas significativas podrían deberse al azar, por lo que se aplicó la corrección de Bonferroni. Esto es, en lugar de valorar una significación de P < 0,01, se consideraron sólo aquellas con una P < (0,01/n), donde n es el número de pruebas realizadas.
Como resultado se obtuvo una hoja de cálculo conteniendo los nombres de los SNPs, el cromosoma donde se encuentran, su posición en el cromosoma, la orientación de las clinas y su significación.
Debido a la particularidad de los resultados obtenidos para el cromosoma Y, se realizó un análisis complementario mediante el programa GenoCline, con el fin de comprobar la verosimilitud de las clinas obtenidas. Este programa implementa el mismo algoritmo para la detección de clinas que GWCline, pero lo realiza sobre frecuencias alélicas, en lugar de sobre genotipos individuales. Para ello se agruparon los individuos en poblaciones y se calcularon las correspondientes frecuencias alélicas.
Tratamiento de las clinas significativas
Una vez obtenidas las clinas, se empleó la base de datos SNP del NCBI para determinar su posible relación con algún gen. De esta forma, se identificaron los SNPs asociados a genes y se procedió a realizar una búsqueda bibliográfica en la base de datos PubMed Central para comprobar si existían evidencias de selección en cada caso.
Resultados
Clinas
Se detectaron clinas con una significación de P<0,01 en un total de 162.417 SNPs. En la Figura 2 se muestra su distribución y orientación. Resulta notable que la mayor parte de las clinas tengan una orientación latitudinal dentro de un rango muy estrecho de valores, ya que la mayor parte se encuentran dentro de un arco de 30 grados.
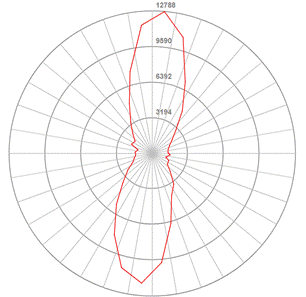
Figura 2. Orientación y frecuencia para arcos de 10º de las clinas detectadas antes de la corrección de Bonferroni.
Figure 2. Orientation and frequency of clines detected before Bonferroni correction, distributed in 10-degree arcs.
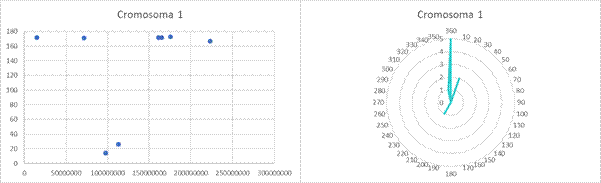
A modo de ejemplo, en la Figura 3 se muestran las posiciones y ángulos de las clinas observadas en el cromosoma 1. Con el fin de facilitar su interpretación, se redistribuyeron las orientaciones para un rango de 0 a 180º, de modo que los valores de 180 a 359º se recalcularon restándoles 180º. Así, las clinas próximas a 0 y 180º son latitudinales y las próximas a 90º son longitudinales. Se observa una mayor densidad de clinas latitudinales. También se observa el espacio causado por el centrómero en el eje de abscisas.
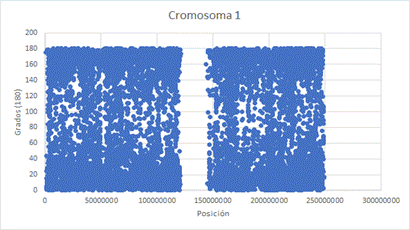
Figura 3. Gráfica de dispersión de la posición en el cromosoma y orientación de los SNPs con p < 0,01 detectados en el Cromosoma 1.
Figure 3. Scatter plot of the position and orientation of SNPs with p < 0.01 on Chromosome 1.
Sin embargo, una gran parte de los resultados significativos podrían deberse al azar, por lo que se aplicó la corrección de Bonferroni. De este modo, solo 292 resultaron ser significativas. En el anexo se muestran las gráficas de dispersión y circulares de cada cromosoma para estas clinas.
Clinas con evidencias de selección
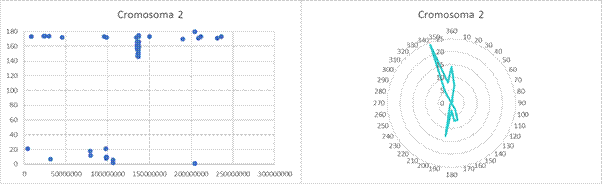
A continuación, se procedió a buscar evidencias de selección en los genes en los que se encontraban los diferentes SNPs. Se encontraron posibles evidencias de selección para un total 37 clinas situadas en 16 genes distintos (Figura 4). La mayoría se encuentran en el cromosoma 2, y los más notables son los genes TMEM163 y LCT, ya que cuentan con el mayor número de SNPs con clinas significativas asociados.
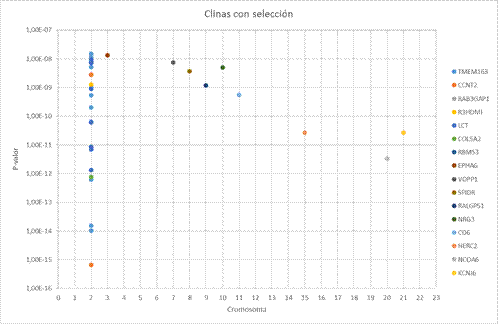
Figura 4. Significación y localización de las clinas con evidencia de selección documentada.
Figure 4. Significance and location of clines with documented evidence of selection.
En el cromosoma 2 encontramos varios genes con evidencias de selección. En primer lugar, el gen LCT es un caso bien estudiado y está relacionado con la persistencia de la actividad lactasa en la edad adulta (Mathieson et al., 2015). En segundo lugar, los genes RAB3GAP1 y R3HDM1 cuentan con un solo SNP cada uno. Son dos genes que se encuentran cerca de la región LCT, y cuentan con evidencias de selección según algunas fuentes (Sabeti et al., 2007; Zhong et al., 2010). En tercer lugar, varias de las clinas que se han considerado producto de la selección están asociadas al gen TMEM163, cuyo alelo ancestral se asocia a la resistencia a la insulina. Esta podría ser la razón por la que ha sido sometido a selección (Sazzini et al., 2016). En cuarto lugar, CCNT2 está relacionado con la interacción entre huésped y patógenos. Los genes con este tipo de funciones comenzaron a presentar una mayor selección desde el periodo neolítico, como resultado de una mayor exposición a agentes patógenos (Wang et al., 2006). En quinto lugar, COL5A2 es un gen relacionado con el colágeno, y algunos genes relacionados con esta proteína han sufrido selección debido a la adaptación al frío en algunas poblaciones de Europa (Harris y DeGiorgio, 2020a).
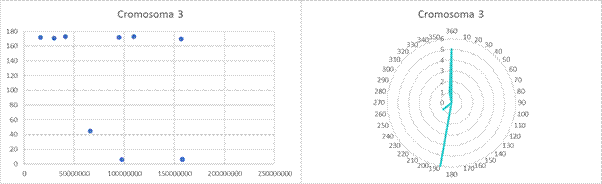
En el cromosoma 3 se han encontrado evidencias de selección en los genes RBMS3 y EPHA6 (Tong et al., 2010; Huber et al., 2016). En el cromosoma 7 existen evidencias de selección en el gen VOPP1 (Kuhlwilm y Boeckx, 2019). En el cromosoma 8 tenemos alguna clina asociada al gen SPIDR, relacionado con la reparación del ADN (Harris y DeGiorgio, 2020b), y en el cromosoma 9 tenemos el gen RALGPS1, relacionado con la pigmentación de la piel (Ju y Mathieson, 2020). En el cromosoma 15 encontramos otro gen relacionado con la pigmentación, HERC2, que afecta a la pigmentación de los ojos (Mathieson et al., 2015) y en el cromosoma 20 presenta evidencias de selección el gen NCOA6 (Sturm y Duffy, 2012).
En el cromosoma 10 el gen NRG3 ha sido detectado como objetivo de selección en la población rusa y un par de muestras europeas del Proyecto 1000 genomas. Este gen está implicado en el desarrollo y el funcionamiento del sistema nervioso, y se ha propuesto que tras la salida de África pudo tener un papel importante en las adaptaciones locales (Khrunin et al., 2020). En el cromosoma 11, CD6 es un gen relacionado con la respuesta inmune (Casadó-Llombart et al., 2021).
Por último, en el cromosoma 21 tenemos el gen KCNJ, con evidencias de haber sido sometido a selección disruptiva en poblaciones italianas (Sazzini et al., 2020).
Clinas sin evidencias de selección
Para el resto de clinas, un total de 255, no se encontraron evidencias de selección. Se puede observar un resumen de su orientación en la Figura 5. A grandes rasgos, prácticamente todas son latitudinales y se encuentran dentro de un arco de 30 grados, salvo las encontradas en el cromosoma Y.
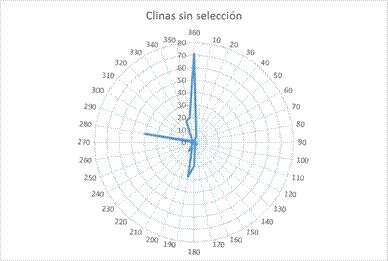
Figura 5. Orientación de las clinas sin evidencia de selección documentada.
Figure 5. Orientation of clines without evidence of selection.
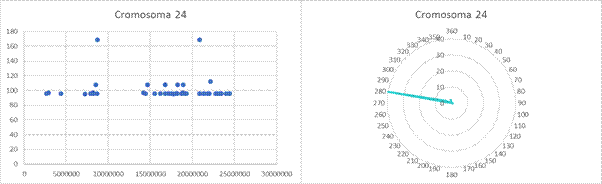
Otro aspecto a destacar es la ausencia de clinas significativas en los cromosomas 14, 22 y 23 tras aplicar la corrección. Además, resulta interesante que, tras descartar las clinas no significativas, siguen predominando las clinas latitudinales, mientras que las longitudinales son minoritarias. Esta tendencia se observa en la mayoría de cromosomas, con la excepción más notable del cromosoma 24 (cromosoma Y).
En relación a los resultados del cromosoma Y, se realizó un nuevo análisis mediante GenoCline. De esta forma, se confirmó la existencia de un grupo de 39 clinas longitudinales con una orientación de 281º virtualmente idénticas, que se ajustan significativamente a una curva sigmoide (Figura 6).
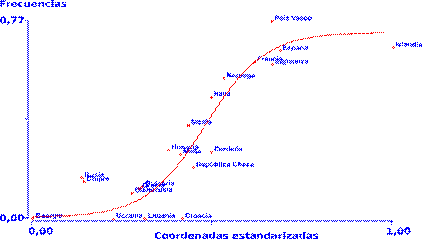
Figura 6. Clina longitudinal del SNP rs7067226 del cromosoma Y. Orientación: 281 grados. Función: y = 0,7221/(1+e**(-12,1343*(x-0,4822)). Pendiente: 12,1343. Punto de inflexión: 0,4822. P (F-ratio): 0,000000. P de los 3 parámetros: (a) 0,000005 (b) 0,002788 (c) 0,000000.
Figure 6. Longitudinal cline of the SNP rs7067226 of the Y chromosome. Orientation: 281 degrees. Function: y = 0.7221/(1+e**(-12.1343*(x-0.4822)). Slope: 12.1343. Inflection point: 0.4822. P (F-ratio): 0.000000. P of the 3 parameters: (a) 0.000005 (b) 0.002788 (c) 0.000000.
Discusión
Clinas con evidencias de selección
Entre las clinas observadas, hay varias asociadas a genes que verosímilmente se encuentran sometidos a presiones selectivas. En primer lugar, en el caso del gen LCT, en Europa se encuentra una frecuencia considerable de persistencia de actividad lactasa desde hace unos 4.000 años (Mathieson et al., 2015). De hecho, se ha sugerido que el alelo responsable de la tolerancia a la lactosa podría haber llegado a Europa por la expansión de los Yamnaya (Allentoft et al., 2015). En poblaciones con un aporte de leche por la existencia de ganadería, ser capaz de digerir la leche sería una ventaja por suponer un aporte seguro de alimento frente a los alimentos cultivados, que se pueden ver mermados de manera súbita. Además, en poblaciones de latitudes altas, en las que la reducida luz solar impide una síntesis suficiente de vitamina D, tendría un gran papel como fuente de este nutriente (Itan et al., 2009).
Los genes RAB3GAP1 y R3HDM1 podrían haber experimentado sus propias presiones selectivas o podrían haberse visto afectados por autoestop genético (Genetic hitchhiking), dada su proximidad al gen LCT.
Es también notable la selección de dos genes relacionados con el sistema inmune, CCNT2 y CD6. Desde el Neolítico, las sociedades humanas tendieron a establecer un modo de vida más sedentario, asociado a un aumento de la densidad poblacional. Esto propició unas tasas de mortalidad elevadas a causa de las epidemias (Peña y Alfonso-Sánchez, 2020). Por tanto, resulta lógico pensar que con la llegada de los granjeros de Anatolia a Europa comenzó una gran presión selectiva sobre genes relacionados con el sistema inmune. La clina relacionada con estos genes podría deberse a una incidencia diferencial de las presiones selectivas causadas por enfermedades (especialmente gastrointestinales) en función de la latitud (Alfonso-Sánchez et al., 2010).
Por otro lado, hay evidencias de selección en tres genes implicados en la pigmentación: HERC2, RALGPS1 y NCOA6. Se cree que el color de piel claro pudo haber comenzado a sufrir una mayor selección desde la transición al Neolítico, ya que se pasó a tener una dieta baja en vitamina D y cualquier reducción en su consumo habría aumentado la selección para un fenotipo de piel clara. En el caso del color de ojos y del pelo, se ha especulado que podría deberse a una interacción con los genes que influyen en el color de la piel, o a algún tipo de selección sexual (Wilde et al., 2014).
Clinas sin evidencias de selección
La selección natural podría explicar algunas de las clinas observadas. Sin embargo, la gran mayoría no se encuentran en genes con evidencias de selección. En este caso, la explicación más probable se encuentra en las diferentes oleadas migratorias ocurridas en el continente europeo desde la entrada de los humanos modernos.
Existen evidencias de una expansión hacia el norte del continente por parte de los cazadores-recolectores del Paleolítico tras el Último Máximo Glacial. En efecto, tras el fin del periodo glacial, se expandieron desde los refugios del sur por el resto del continente. Esta expansión ha sido respaldada por datos genéticos y arqueológicos (Torroni et al., 1998).
La entrada de la agricultura en Europa ocurrió por el sudeste del continente, desde Anatolia, y se extendió en dirección norte y en dirección oeste (Omrak et al., 2016; Skoglund et al., 2012).
También se ha constatado la existencia de una migración desde la Estepa Póntica hacia el sudoeste de Europa durante la Edad de Bronce (Jones et al., 2015; Allentoft et al., 2015).
En conjunto, la recolonización postglacial del Paleolítico desde el sur del continente, la difusión démica del Neolítico desde el sudeste y la oleada de los hombres de las estepas desde el nordeste, podrían justificar un vector resultante de componente mayoritariamente latitudinal en las clinas detectadas, si bien cabría esperar un arco más amplio de direcciones para el conjunto de las clinas.
Por el contrario, resulta sorprendente el caso del cromosoma Y. Se han encontrado un total de 48 SNPs, de los que 39 han mostrado clinas virtualmente idénticas de tipo longitudinal. Este conjunto de SNPs (ligados, pues se encuentran en la región no recombinante del cromosoma Y) configura un haplotipo que se identifica con el haplogrupo R1b-M269 (R1b1a1b), el más frecuente en Europa y cuya distribución es conocida; en efecto, se observa una mayor frecuencia de este haplotipo en el oeste de Europa (Myres et al., 2011) y según algunos estudios se ha asociado a la expansión Yamnaya (Olalde et al., 2018). Sin embargo, resulta curioso que sea muy poco frecuente en las regiones del norte y nordeste de Europa, donde la influencia de los hombres de las estepas es más evidente.
El resto de haplogrupos frecuentes en Europa, particularmente en su mitad oriental, no han revelado clinas. Es el caso de a) R1a, asociado también con la expansión de las lenguas indoeuropeas, b) I, con un origen posible en el Paleolítico europeo, c) E1b1b y J, presumiblemente introducidos durante el Neolítico, y d) G y N, posiblemente de origen asiático.
Por otra parte, las clinas observadas para R1b-M269 son de tipo sigmoide, apuntando a la existencia de dos grupos poblacionales de diferente composición, con una zona de contacto en una franja central que vendría definida de nornoroeste a sursudeste por Noruega, la República Checa, Hungría e Italia.
Conclusiones
En vista de los resultados obtenidos en este trabajo, se pueden enunciar una serie de conclusiones. En primer lugar, la mayoría de las clinas detectadas en el genoma de los europeos actuales son latitudinales, con algunas clinas longitudinales presentes de manera minoritaria. Las clinas latitudinales probablemente sean el resultado de las tres principales oleadas migratorias (recolonización postglacial del Paleolítico, difusión del Neolítico y expansión Yamnaya).
En segundo lugar, una vez aplicada la corrección de Bonferroni prácticamente no se ha detectado la presencia de clinas latitudinales en el cromosoma Y, sino que básicamente hay una clina longitudinal en un haplotipo configurado por 39 SNPs, correspondiente al haplogrupo R1b-M269. A pesar de su orientación, se ha asociado a la expansión Yamnaya.
El estudio del genoma europeo es una gran fuente de información sobre el pasado de sus poblaciones, y las clinas pueden ser un buen reflejo del resultado de las grandes oleadas migratorias. Su análisis puede suponer un aporte enriquecedor para comprender la composición del patrimonio genético de las poblaciones humanas actuales.
Bibliografía
Alfonso-Sánchez, M. A., Pérez-Miranda, A. M., García-Obregón, S., Peña, J. A. 2010. An evolutionary approach to the high frequency of the Delta F508 CFTR mutation in European populations. Medical hypotheses, 74(6), 989-992. doi: 10.1016/j.mehy.2009.12.018.
Allentoft, M.E., Sikora, M., Sjögren, K., et al. (2015). Population genomics of Bronze Age Eurasia. 2015. Nature, 522, 167–172. doi: 10.1038/nature14507.
Casadó-Llombart, S., Velasco-de Andrés, M., Català, C., Leyton-Pereira, A., Lozano, F. y Bosch, E. 2021. Contribution of Evolutionary Selected Immune Gene Polymorphism to Immune-Related Disorders: The Case of Lymphocyte Scavenger Receptors CD5 and CD6. International Journal of Molecular Sciences, 22(10), 5315. doi:10.3390/ijms22105315.
Cavalli-Sforza, L.L. 2005. The Human Genome Diversity Project: past, present and future. Nature Reviews Genetics, 6, 333–340. doi:10.1038/nrg1596.
Fu, Q., Posth, C., Hajdinjak, M., et al. 2016. The genetic history of Ice Age Europe. Nature, 534, 200–205. doi: 10.1038/nature17993.
Gokcumen, O. y Frachetti, M. 2020. The Impact of Ancient Genome Studies in Archaeology. Annual Review of Anthropology, 49, 277–298. doi:10.1146/annurev-anthro-010220-074353.
Haak, H., Lazaridis, I., Patterson, N., et al. 2015. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211. doi: 10.1038/nature14317.
Harris, A.M. y DeGiorgio, M. 2020a. A Likelihood Approach for Uncovering Selective Sweep Signatures from Haplotype Data. Molecular biology and evolution, 37(10), 3023–3046. doi:10.1093/molbev/msaa115.
Harris, A.M. y DeGiorgio, M. 2020b. Identifying and Classifying Shared Selective Sweeps from Multilocus Data. Genetics 215, 143–171. doi:10.1534/genetics.120.303137.
Hofmanová, Z., Kreutzer, S., Hellenthal, G., et al. 2016. Early farmers from across Europe directly descended from Neolithic Aegeans. PNAS, 113(25), 6886–6891. doi: 10.1073/pnas.1523951113.
Huber, C.D., Degiorgio, M., Hellmann, I.S. y Nielsen, R. 2016. Detecting recent selective sweeps while controlling for mutation rate and background selection. Molecular Ecology, 25(1), 142–156. doi:10.1111/mec.13351.
Isern, M., Fort, J. y de Rioja, V.L. (2017). The ancient cline of haplogroup K implies that the Neolithic transition in Europe was mainly demic. Scientific Reports, 7(1), 11229. doi:10.1038/s41598-017-11629-8.
Itan, Y., Powell, A., Beaumont, M.A., Burger, J. y Thomas, M.G. 2009. The Origins of Lactase Persistence in Europe. PLoS Comput Biol, 5(8), e1000491. doi: 10.1371/journal.pcbi.1000491.
Jones, E.R., Gonzalez-Fortes, G., Connell, S., et al. 2015. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nature Communications 6, 8912. doi: 10.1038/ncomms9912.
Jones, E.R., Zarina, G., Moiseyev, V., et al. 2017. The Neolithic Transition in the Baltic Was Not Driven by Admixture with Early European Farmers. Current biology, 27(4), 576–582. doi:10.1016/j.cub.2016.12.060.
Ju, D. y Mathieson, I. 2020. The evolution of skin pigmentation-associated variation in West Eurasia. Proceedings of the National Academy of Sciences of the United States of America, 118(1), e2009227118. doi: 10.1073/pnas.2009227118.
Khrunin, A.V., Khvorykh, G.V., Fedorov, A.N., Limborska, S.A. 2020. Genomic landscape of the signals of positive natural selection in populations of Northern Eurasia: A view from Northern Russia. PloS one, 15(2), e0228778. doi:10.1371/journal.pone.0228778.
Kuhlwilm, M. y Boeckx, C. 2019. A catalog of single nucleotide changes distinguishing modern humans from archaic hominins. Scientific reports, 9(1), 8463. doi:10.1038/s41598-019-44877-x.
Lawson-Handley, L.J., Manica , A., Goudet, J. y Balloux, F. 2007. Going the distance: human population genetics in a clinal world. Trends in Genetics, 23(9), 432–439. doi:10.1016/j.tig.2007.07.002.
Lazaridis, I., Nadel, D., Rollefson, G., et al. 2016. Genomic insights into the origin of farming in the ancient Near East. Nature, 536, 419–424. doi:10.1038/nature19310.
Lazaridis, I., Patterson, N., Mittnik, A., et al. 2014. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature, 513, 409–413. doi:10.1038/nature13673.
Mathieson, I., Lazaridis, I., Rohland, N., et al. 2015. Genome-wide patterns of selection in 230 ancient Eurasians. Nature, 528, 499–503. doi: 10.1038/nature16152.
Myres, N.M., Rootsi, S., Lin, A.A., et al. 2011. A major Y-chromosome haplogroup R1b Holocene era founder effect in Central and Western Europe. European Journal of Human Genetics, 19, 95–101. doi: 10.1038/ejhg.2010.146.
Olalde, I., Brace, S., Allentoft, M.E., et al. 2018. The Beaker phenomenon and the genomic transformation of northwest Europe. Nature, 555, 190–196. doi: 10.1038/nature25738.
Olalde, I., Mallick, S., Patterson, N., et al. 2019. The genomic history of the Iberian Peninsula over the past 8000 years. Science, 363(6432), 1230–1234. doi: 10.1126/science.aav4040.
Omrak, A., Günther, T., Valdiosera, C., et al. 2016. Genomic Evidence Establishes Anatolia as the Source of the European Neolithic Gene Pool. Current Biology, 26(2), 270–275. doi: 10.1016/j.cub.2015.12.019.
Peña, J.A. 2022. GWCline. Comunicación personal.
Peña, J.A. y Alfonso-Sánchez, M.A. 2020. Crisis de mortalidad en dos poblaciones rurales del País Vasco durante los siglos XIX y XX. Revista Española de Antropología Física, 42, 24–31.
Peña, J. A., Gómez-Pérez, L., y Alfonso-Sánchez, M. A. 2022. On the Trail of Spatial Patterns of Genetic Variation. Evolutionary Biology, 49(1), 84-91. doi: 10.1007/s11692-021-09552-y.
Pinhasi, R., Thomas, M.G., Hofreiter, M., Currat, M. y Burger, J. 2012. The genetic history of Europeans. Trends in Genetics, 28(10), 496–505. doi:10.1016/j.tig.2012.06.006.
Rosser, Z.H., Zerjal, T., Hurles, M.E., et al. 2000. Y-Chromosomal Diversity in Europe Is Clinal and Influenced Primarily by Geography, Rather than by Language. The American Journal of Human Genetics, 67(6), 1526–1543. doi:10.1086/316890.
Saag, L., Vasilyev, S.V., Varul, L., et al. 2021. Genetic ancestry changes in Stone to Bronze Age transition in the East European plain. Science advances, 7(4), eabd6535. doi: 10.1126/sciadv.abd6535.
Sabeti, P.C., Varilly, P., Fry, B., et al. 2007. Genome-wide detection and characterization of positive selection in human populations. Nature, 449(7164), 913–918. doi:10.1038/nature06250.
Sazzini, M., Abondio, P., Sarno, S., et al. 2020. Genomic history of the Italian population recapitulates key evolutionary dynamics of both Continental and Southern Europeans. BMC Biology, 18, 51. doi:10.1186/s12915-020-00778-4.
Sazzini, M., Gnecchi-Ruscone, G.A., Giuliani, C., et al. 2016. Complex interplay between neutral and adaptive evolution shaped differential genomic background and disease susceptibility along the Italian peninsula. Scientific reports, 6, 32513. doi: 10.1186/s12915-020-00778-4.
Skoglund, P., Malmström, H., Raghavan, M., et al. 2012. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science, 336(6080), 466–469. doi: 10.1126/science.1216304.
Skoglund, P. y Mathieson, I. 2018. Ancient Genomics of Modern Humans: The First Decade. Annual Review of Genomics and Human Genetics, 19, 381–404. doi:10.1146/annurev-genom-083117-021749.
Sturm, R.A., y Duffy, D.L. 2012. Human pigmentation genes under environmental selection. Genome biology, 13(9), 248. doi: 10.1186/gb-2012-13-9-248.
Tong, P., Prendergast, J.G., Lohan, A.J., et al. 2010. Sequencing and analysis of an Irish human genome. Genome biology, 11(9), R91. doi:10.1186/gb-2010-11-9-r91.
Torroni, A., Bandelt, H., D’Urbano, L., Lahermo, P., Moral, P. 1998. mtDNA Analysis Reveals a Major Late Paleolithic Population Expansion from Southwestern to Northeastern Europe. American Journal of Human Genetics, 62, 1137–1152. doi: 10.1086/301822.
Wang, E.T., Kodama, G., Baldi, P., y Moyzis, R.K. 2006. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proceedings of the National Academy of Sciences of the United States of America, 103(1), 135–140. doi: 10.1073/pnas.0509691102.
Wilde, S., Timpson, A., Kirsanow, K., et al. 2014. Direct evidence for positive selection of skin, hair, and eye pigmentation in Europeans during the last 5,000 y. PNAS, 111(13) 4832–4837. doi: 10.1073/pnas.1316513111.
Zhong, M., Lange,
K., Papp, J.C. y Fan, R. 2010. A powerful score test to detect positive
selection in genome-wide scans. European Journal of Human Genetics 18,
1148–1159. doi:10.1038/ejhg.2010.60.
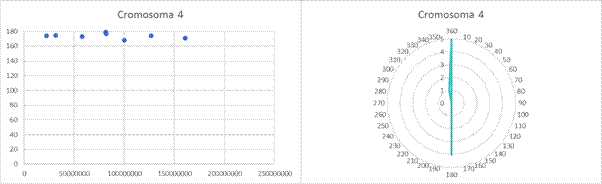
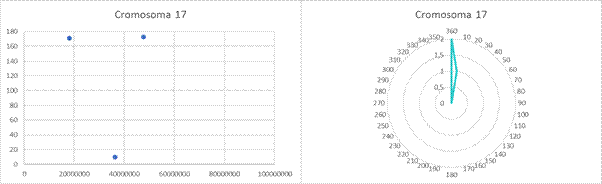
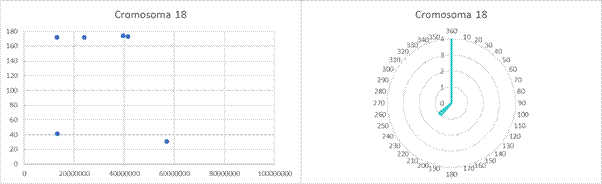
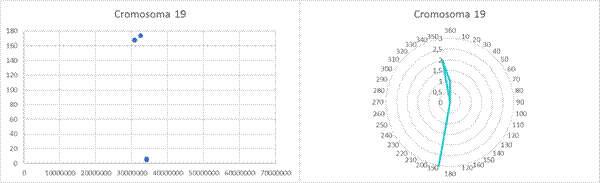
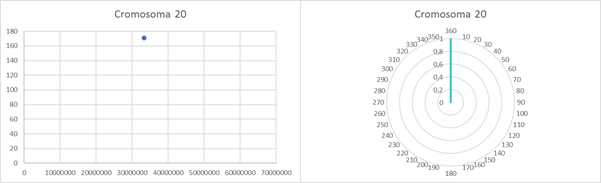
Anexo
Gráficas de dispersión y gráficas circulares de las clinas observadas en cada cromosoma tras aplicar la corrección de Bonferroni.
Scatter plots and pie charts of the clines observed in each chromosome after applying the Bonferroni correction.