
Peña, J.A., Izagirre, N., Rebato, E., 2019. La filogenia Primate: Un protocolo de prácticas. Antropo, 42, 55-61. www.didac.ehu.es/antropo
La filogenia Primate: Un protocolo de prácticas
Primate Phylogeny: A Practice Protocol
Jose A. Peña, Neskuts Izagirre, Esther Rebato
Departamento de Genética, Antropología Física y Fisiología Animal. Facultad de Ciencia y Tecnología. Universidad del País Vasco (UPV/EHU). España
Correspondencia: Jose A. Peña. joseangel.pena@ehu.eus
Palabras clave: Protocolo de prácticas, Genómica Primate, Filogenia, MEGA, GEH.
Keywords: Practice protocol, Primate Genomics, Phylogeny, MEGA, GEH.
Resumen
Objetivo: Se propone la impartición de una clase práctica de análisis de datos genómicos, en la que se revisa la filogenia del orden de los primates.
Materiales y métodos: A partir de las secuencias de 54 genes correspondientes a 186 especies de primates se realizan una serie de análisis filogenéticos mediante diferentes algoritmos. En primer lugar, se comparan los resultados obtenidos mediante cinco métodos (Máxima Verosimilitud, Unión de Vecinos, Evolución Mínima, UPGMA y Máxima Parsimonia) para el conjunto del orden Primate. Después, se profundiza en el análisis por separado de Strepsirrhini y Haplorrhini (Platyrrhini y Catarrhini). Con el fin de facilitar la identificación visual de los diferentes géneros de primates, se ha desarrollado un programa (GEH), que muestra imágenes tanto de cada especimen como de su distribución geográfica.
Actividades: En el informe final de la actividad se valorarán los algoritmos filogenéticos, seleccionándose el método que ofrezca el árbol más verosímil a partir de los conocimientos adquiridos previamente sobre los diferentes grupos de primates. Se valorará asimismo la robustez de las diferentes ramas a la luz de los valores bootstrap obtenidos. En los análisis de Strepsirrhini, Platyrrhini y Catarrhini, se interpretarán las agrupaciones de géneros en los respectivos árboles. Finalmente, se discutirán las características morfológicas diferenciales más llamativas y la distribución geográfica de cada grupo.
Abstract
Objective: A practical class on genomic data analysis is proposed, in which phylogeny of the order of primates is reviewed.
Materials and methods: From the sequences of 54 genes corresponding to 186 primate species, a series of phylogenetic analyses are carried out using different algorithms. First, the results obtained by five methods (Maximum Likelihood, Neighbor Joining, Minimum Evolution, UPGMA and Maximum Parsimony) are compared for the whole order Primate. Then, the analysis of Strepsirrhini and Haplorrhini (Platyrrhini and Catarrhini) is deepened separately. In order to facilitate the visual identification of the different primate genera, a program (GEH) has been developed, which shows images of both each specimen and its geographical distribution.
Activities: In the final report of the activity, the phylogenetic algorithms will be evaluated, selecting the method that offers the most credible tree from the knowledge previously acquired about the different groups of primates. The robustness of the different branches will also be assessed in the light of the bootstrap values obtained. In the Strepsirrhini, Platyrrhini and Catarrhini analyses, the genus groupings in the respective trees will be interpreted. Finally, the most striking differential morphological characteristics and geographical distribution of each group will be discussed.
El Orden Primates
El orden de los primates (Linneo, 1758) se caracteriza por ser un grupo de mamíferos poco especializados, lo cual dificulta su definición, ya que algunas de sus características son comunes a los otros órdenes de mamíferos placentarios. Los primates surgieron a finales del Cretácico, hace unos 85-55 millones de años a partir de pequeños mamíferos terrestres. Su distribución actual (con algunas excepciones y sin tener en cuenta a los humanos) de sitúa en la franja comprendida entre los 25ºN y los 30ºS, es decir, entre los trópicos de Cáncer y Capricornio; una distribución más reducida que la presentada por este orden en épocas anteriores y que no incluye a Europa (salvo Gibraltar), Oceanía o la Antártida. En su conjunto, los primates presentan una gran variedad de formas que muestran diferencias anatómicas, fisiológicas, morfológicas y de comportamiento, relacionadas en general con su tipo de hábitat, principalmente arborícola en medio tropical (selvas, sabanas, praderas, pantanos, manglares y diversos tipos de bosques). En la actualidad, se reconocen al menos 455 especies de primates, 75 géneros y 16 familias (Groves, 2005), y se han descrito 11 nuevas especies desde 2010.
En la cladística actual se considera que los primates son un orden monofilético que forma parte del clado Euarchontoglires o “Supraprimates”. Los estudios genéticos indican una estrecha relación de los primates con los colugos (orden Dermoptera) y los tupayas (orden Scadentia). Los tres órdenes configuran el clado Euarchonta, que, según numerosas evidencias moleculares, estaría estrechamente relacionado con el clado de los Glires al que pertenecen los órdenes Rodentia (roedores) y Lagomorpha (conejos y otros). Siempre ha existido una cierta dificultad en la terminología aplicada a los primates y sobre todo en las clasificaciones que son numerosas y diversas y no siempre aceptadas por todos los primatólogos. Gran parte de los problemas que subyacen en la clasificación de los primates radica en los caracteres utilizados para su organización en grupos taxonómicos (suborden, infraorden, superfamilia, familia, género), lo cual dificulta también el estudio de su evolución y relaciones filogenéticas. Es evidente que la sistemática es una disciplina muy activa en la que existen numerosos debates y discrepancias y como indica Gommery (2005), sólo la especie, el género y la familia tienen la suficiente entidad para el código de nomenclatura. Las clasificaciones actuales de los cladistas, basadas no sólo en los rasgos anatómicos, sino cada vez más en datos moleculares, tienden en algunos casos a multiplicar las categorías taxonómicas y algunas de estas clasificaciones pueden no adaptarse por ejemplo a las de los primates ya extintos.
Filogenia de los primates
La filogenia trata de reconstruir las relaciones evolutivas entre diferentes especies (u otras entidades como los genes, las poblaciones, etc.), asumiendo que todas ellas descienden de ancestros comunes más o menos alejados en el tiempo mediante sucesivas bifurcaciones (aunque se sabe hoy día que también existen fusiones) y partiendo de esta premisa, intenta determinar cuáles son las distancias genéticas o los tiempos de separación entre estas especies. Para ello, considera diversas evidencias, tales como los datos morfológicos, los genotipos o las secuencias de ADN o de proteínas, entre otras. Actualmente, las relaciones filogenéticas existentes entre los primates actuales se están investigando de forma exhaustiva, utilizando para ello secuencias de nucleótidos de diferentes genomas (nucleares y mitocondriales) y otros tipos de marcadores como los SINEs (p.e. en los Monos del Nuevo Mundo o Platirrinos, ver Opazo et al. 2006), aunque los resultados a veces no son concordantes, pues dependen, por ejemplo, del tipo de marcadores utilizados.
El campo de la genómica comparada se encuentra hoy día en una rápida expansión y está proporcionando herramientas para la reconstrucción de filogenias, en particular la del orden de los primates, incluida nuestra especie que comparte un ancestro común con otros primates no humanos. Así, la secuenciación primero del genoma humano completo (2001, 2004) y posteriormente la del chimpancé (Pan troglodytes) en 2005 y otros grandes simios como el orangután de Sumatra (Pongo abelii) en 2011, el gorila (Gorilla gorilla) y el bonobo (Pan paniscus) en 2012, además de otros primates (p.e. Macaca mulatta en 2007 y Macaca fascicularis en 2011) o el Gibón (Nomascus leucogenys) en 2014, ha posibilitado la comparación de homologías, sintenias y pseudogenes compartidos en el genoma de las distintas especies. Es innegable que el proyecto genoma humano ha revolucionado el campo de la genómica, la proteómica y la medicina. El interés de la genómica comparativa de los primates radica en que i) permite reconstruir los mecanismos de cambio genómico, en particular la actuación de la selección natural direccional o estabilizadora (ver McVicker et al. 2009; Enard et al. 2010), sobre todo aquellos sucesos adaptativos que expliquen las particularidades biológicas que nos hacen humanos, ii) abre nuevas perspectivas en el conocimiento del proceso de especiación y divergencia genética entre las líneas evolutivas de los humanos y de los grandes simios, así como la reconstrucción de filogenias (los datos genómicos permiten ensamblar alineaciones que aumentan considerablemente el número de sitios informativos para el análisis de las fechas de divergencia molecular), la dinámica macroevolutiva y la biogeografía histórica de la diversificación de los primates (ver Steiper y Young, 2006; Springer et al. 2012) y iii) considera a los primates como modelo para el estudio de enfermedades humanas (aplicaciones evolutivas en un contexto biomédico) y la predicción funcional de variantes genéticas en nuestra especie (ver Rogers y Gibbs, 2014).
Como señalan Perelman et al. (2011) en su propuesta de una filogenia molecular para los primates actuales “los análisis genómicos comparativos de primates ofrecen un potencial considerable para definir y comprender los procesos que moldean, configuran y transforman el genoma humano”. Sin embargo, como ya se ha señalado anteriormente, la taxonomía de los primates es a la vez compleja y controvertida, y no hay un consenso unificado sobre la jerarquía evolutiva de las especies de primates existentes en la actualidad. La filogenia molecular propuesta por los autores mencionados es muy consistente e “ilumina los eventos de la evolución de los primates desde la antigüedad hasta la actualidad, aclarando numerosas controversias taxonómicas y proporcionando nuevos datos sobre la evolución humana” (Perelman et al. 2011). Este interesante trabajo revela que los genomas de los primates muestran notables diferencias en los patrones de especiación, en la diversidad genómica, en las tasas de evolución y en las frecuencias de los eventos de inserción/deleción. En definitiva, el análisis de las relaciones filogenéticas y los tiempos de divergencia entre las distintas especies de primates son cruciales para comprender la historia evolutiva y biogeográfica de este orden de mamíferos, incluyendo los patrones de diversificación en relación con los cambios ambientales (Springer et al. 2012).
En este artículo se proponen una serie de actividades que, en una extensión de tiempo limitada a unas 2 o 3 horas, permiten una primera aproximación al conocimiento de la filogenia de los primates y de los géneros y especies que la componen.
Materiales
Se propone la elaboración de una serie de árboles filogenéticos. Con este fin, se utilizará la base de datos de Perelman et al (2011), que comprende la secuencia de 54 genes de 186 especies de primates, distribuídas en 61 géneros diferentes, lo que supone aproximadamente el 90% de los géneros de este orden. Se incluyen además un género de Scandentia (Tupaia), dos de Dermoptera (Cynocephalus y Galeopterus) y uno de Lagomorpha (Oryctolagus), con el fin de disponer de raíz en los árboles filogenéticos.
Esta base de datos se ha sudividido, mediante NDE (Nexus Data Editor) en:
- DBPrimateGeneros.mega. Incluye todos los géneros de primates, representados por una sola especie cada uno. Son 61 secuencias, además de los especímenes no primates.
- DBStrepsirrhini.mega. Comprende las 42 especies disponibles de Strepsirrhini.
- DBSPlatyrrhini.mega. Contiene 64 especies de platirrinos.
- DBSCatarrhini.mega. Incluye 78 especies de catarrinos.
Los árboles filogenéticos se realizarán con el programa Mega-X (Kumar et al, 2018).
Como apoyo a la interpretación de las filogenias, se ha desarrollado el programa GEH, que muestra imágenes obtenidas de Wikipedia de diferentes especímenes de primates y de su distribución geográfica.
Protocolo
Orden Primate
Para trabajar con el conjunto del orden Primate y con el fin de facilitar la visualización de los árboles, se ha limitado el análisis a una especie por género. La base de datos utilizada se denomina DBPrimateGeneros.mega.
Se inicia MEGA-X (Figura 1). Se abre el fichero de datos, <File><Open a File/Session>.
Si se selecciona <Data><Explore Active Data>, se puede ver el listado de especies y sus secuencias.
Figura 1. Pantalla de inicio de MEGA-X.
Figure 1. MEGA-X startup screen.
Se van a probar los cinco métodos diferentes disponibles en Mega para la realización de árboles filogenéticos (Figura 2).
Cinco algoritmos
Se comienza haciendo un árbol filogenético mediante el método de Máxima Verosimilitud, <Phylogeny><Construct/Test Maximum Likelihood Tree>.
Una vez concluídos los cálculos, aparece una nueva ventana con el árbol obtenido.
Se indica la raíz del árbol (rabbit) y se rota. Para ello, se señala la rama correspondiente al conejo y después <Subtree><Root>. Se salva la imagen del árbol, <Image><Save as Png file>.
En segundo lugar, se construye un árbol filogenético mediante el método de Unión de Vecinos, <Phylogeny><Construct/Test Neighbor Joining Tree>. Se aceptan las opciones predeterminadas. Se selecciona la raíz del árbol y se rota. De nuevo, se guarda la imagen del árbol.
A continuación, se dibuja un árbol filogenético mediante el método de Evolución Mínima, <Phylogeny><Construct/Test Minimum Evolution Tree>. Se procede como en los casos anteriores.
El cuarto árbol se obtendrá mediante el método UPGMA <Phylogeny><Construct/Test UPGMA Tree>.
Por último, se realizará un árbol filogenético mediante el método de Máxima Parsimonia, <Phylogeny><Construct/Test Maximum Parsimony Tree>.
Puede encontrarse una interesante comparación de varios de estos métodos en Saitou e Imanishi (1989).
Figura 2. Métodos para la elaboración de árboles filogenéticos
de MEGA-X.
Figure 2. Methods of phylogenetic tree construction with MEGA-X.
Actividad propuesta
Se propone la comparación de los cinco árboles obtenidos, analizando sus diferencias y valorando la coherencia de cada uno a la luz de nuestros conocimientos sobre la taxonomía primate.
Bootstrap
Con el fin de contrastar la robustez de las diferentes ramas de un árbol, puede realizarse un análisis bootstrap. Puesto que en algunos casos Mega puede tardar un tiempo considerable en hacerlo, se probará únicamente sobre el árbol obtenido mediante Máxima Parsimonia. <Phylogeny><Construct/Test Maximum Parsimony Tree>. Se seleccionará <Phylogeny Test/Test of Phylogeny/Bootstrap Method> con 100 repeticiones <OK>.
Actividad propuesta
Se propone interpretar los valores bootstrap de la filogenia primate obtenida mediante Máxima Parsimonia.
Strepsirrhini
A continuación se realizará un árbol considerando las 42 especies disponibles de Strepsirrhini. Para ello, se utilizará el método seleccionado en el apartado anterior.
La base de datos a utilizar es DBStrepsirrhini.mega.
Haplorrhini
Platyrrhini
Utilizando la base de datos DBSPlatyrrhini.mega, se realizará un nuevo árbol sobre un grupo de 64 especies de platirrinos.
Catarrhini
Por último, se realizará un análisis filogenético a partir de la base de datos DBSCatarrhini.mega, que incluye 78 especies de catarrinos.
Actividad propuesta
Se propone valorar la coherencia de la agrupación de las diferentes especies de cada género y las agrupaciones de géneros en los tres últimos árboles.
Características morfológicas y distribución geográfica
Mediante el programa GEH, desarrollado para esta práctica, se revisarán una serie de fotografías de diferentes especies de primates, junto a los mapas en los que se describe su distribución geográfica (Figura 3).
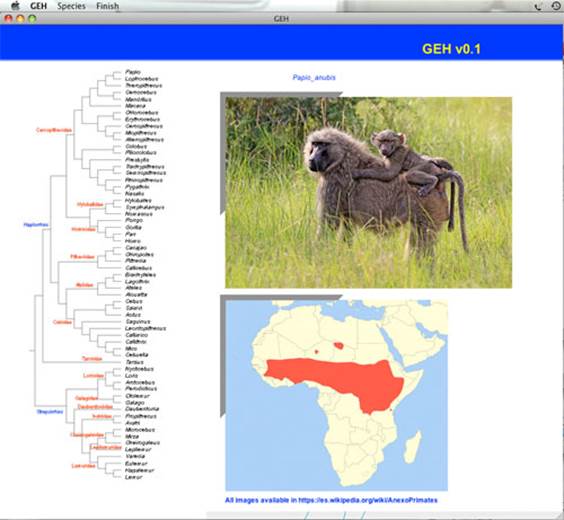
Figura 3. Filogenia Primate, e imágenes mostradas por el
programa GEH.
Figure 3. Primate phylogeny, and images shown by the GEH program.
Actividad propuesta
Se revisará y comentará la distribución geográfica de Strepsirrhini y Haplorrhini (distinguiendo entre Platyrrhini y Catarrhini). Asimismo, se describirán las principales características en cada grupo del rinario, la posición de los orificios nasales y la existencia de cola.
Enlaces
Listado de genes secuenciados.
DBPrimateGeneros.mega.
http://www.ehu.eus/~ggppegaj/EvolHum/DBs/DBPrimateGeneros.mega
DBStrepsirrhini.mega.
http://www.ehu.eus/~ggppegaj/EvolHum/DBs/DBStrepsirrhini.mega
DBSPlatyrrhini.mega.
http://www.ehu.eus/~ggppegaj/EvolHum/DBs/DBPlatyrrhini.mega
DBSCatarrhini.mega.
http://www.ehu.eus/~ggppegaj/EvolHum/DBs/DBCatarrhini.mega
NDE.
https://www.softpedia.com/get/Science-CAD/NEXUS-Data-Editor.shtml
MegaX win64.
GEH.
http://www.ehu.eus/~ggppegaj/Software/GEH.zip
Bibliografía
Enard, D., Depaulis, F., Crollius, H., 2010, Human and Non-Human Primate Genomes Share Hotspots of Positive Selection. PLoS Genetics, 6(2): e1000840. doi:10.1371/journal.pgen.1000840.
Gommery, D., 2005, Evolución de los Primates. En Para comprender la Antropología Biológica. Evolución y Biología humana, editado por E. Rebato, C. Susanne y B. Chiarelli (EDV, Estella, Navarra) p. 221.
Groves, C.P., 2005, Order Primates. En Mammal species of the World, Third Edition. Editado por D.E. Wilson y D.M. Reeder (The Johns Hopkins University Press, Baltimore) p. 111.
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular biology and evolution, 35(6), 1547-1549.
Linneo, C., 1758, Systema naturæ: per regna tria naturaæ, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis. Tomo 1. Editio Decima Reformata. (Holmiæ, Estocolmo: Impensis Direct Laurentii Salvii. Disponible en Biodiversitas Heritage Library) p. 824. doi:10.5962/bhl.title.542.
McVicker, G., Gordon, D., Davis, C., Green, P., 2009, Widespread Genomic Signatures of Natural Selection in Hominid Evolution. PLoS Genetics, 5(5): e1000471. doi:10.1371/journal.pgen.1000471.
Opazo, J.C., Wildman, D.E., Prychitko, T., Johnson, R.M., 2006, Phylogenetic relationships and divergence times among New World monkeys (Platyrrhini, Primates). Molecular Phylogenetics and Evolution 40: 274-280.
Perelman, P., Johnson, W.E., Roos, C., Seuánez, H.N., Horvath, J.E., Moreira, M.A.M., Kessing, B., Pontius, J., Roelke, M., Rumpler, Y., Schneider, M.P.C., Silva, A., O’Brien, S.J., Peccon-Slattery, J., 2011, A Molecular Phylogeny of Living Primates. PLoS Genetics, 7(3): e1001342. doi:10.1371/journal.pgen.1001342.
Rogers, J., Gibbs, R.A., 2014. Comparative primate genomics: emerging patterns of genome content and dynamics. Nature Reviews, 15: 347-359.
Saitou, N., Imanishi, T. 1989. Relative efficiencies of the Fitch-Margoliash, maximum-parsimony, maximum-likelihood, minimum-evolution, and neighbor-joining methods of phylogenetic tree construction in obtaining the correct tree. Mol. Bio. Evol. 6, 514-525.
Springer, M.S., Meredith, R.W., Gatesy, J., Emerling, C.A., Park, J., Rabosky, D.L., Stadler, T., Steiner, C., Ryder, O.A., Janecka, J.E., Fisher, C.A., Murphy, W.J., 2012, Macroevolutionary Dynamics and Historical Biogeography of Primate Diversification Inferred from a Species Supermatrix. PLoS ONE, 7(11): e49521. doi:10.1371/journal.pone.0049521.
Steiper, M.E., Young, N.M., 2006, Primate molecular divergence dates. Molecular Phylogenetics and Evolution, 41: 384-394.