
Hamzi, K., Bellayou, H., Slassi, I., Nadifi, S., 2010,
La maladie de Steinert dans une famille marocaine: phénomène d’anticipation et conseil génétique. Antropo, 23,
73-76. www.didac.ehu.es/antropo
La
maladie de Steinert dans une famille marocaine:
phénomène d’anticipation et
conseil génétique
Steinert’s
disease in a moroccan family: Phenomenon
of anticipation and genetic counseling
K. Hamzi, H. Bellayou, I. Slassi, S. Nadifi
Laboratoire de
Génétique Médicale et Pathologie Moléculaire (LGPM), Faculté de Médecine et de
Pharmacie- Casablanca; 19, rue Tarik- Ibn- Ziad, B.P. 9154, 10 000 Casablanca,
Maroc.
Auteur chargé de
la correspondance: Khalil
HAMZI, Laboratoire de Génétique Médicale et Pathologie Moléculaire (LGPM),
Faculté de Médecine et de Pharmacie- Casablanca; 19, rue Tarik- Ibn- Ziad, B.P.
9154, 10 000 Casablanca, Maroc. E-mail: khalil.hamzi@yahoo.fr
Mots clés : conseil génétique, maladie de Steinert, phénomène anticipation, surveillance.
Key
words: genetic counseling, phenomenon of anticipation,
Steinert’s disease, surveillance.
Résumé
La maladie de Steinert est la dystrophie musculaire la plus fréquente. Elle se caractérise par une atteinte multiviscérale qui définit 4 formes cliniques partant de la forme bénigne de l’adulte à la forme néonatale d’une extrême gravité. Le gène responsable de la DM1 (DMPK) localisé sur la région 19q13.3, se caractérise par des répétitions CTG dont le nombre, à l’état normal, varie de 5 à 35 répétitions et à l’état morbide passe de 50 à plus 3000 répétitions.
La maladie de Steinert est caractérisée par une hétérogénéité clinique ce qui pose le problème diagnostique, nécessitant le recours à des méthodes de TP-PCR et de Southern-blot pour la confirmation diagnostique.
Le but de notre travail est de confirmer, dans un premier temps, le diagnostic de DM1 chez les membres d’une famille présentant un tableau clinique évoquant à priori une maladie de Steinert avec aggravation clinique à travers les 3 générations et proposer dans un deuxième temps un conseil génétique approprié.
Abstract
Steinert's disease or DM1 is the most frequent muscular dystrophy. It’s characterized by systemic attacks defines 4 clinical forms. The gene responsible of DM1 (DMPK)
is located on the 19q13.3 region, is characterized by CTG repeats, normal
number varies between 5 and 35 repetitions and the morbid allele from 50 to
more 3000 repetitions.
Steinert's disease is characterized by clinical heterogeneity and this
raises diagnosis problem, requiring the use of methods of TP-PCR and Southern
blot to confirm the diagnosis.
The aim of our work is to confirm the diagnosis of DM1 in a Moroccan
family with a clinical picture evoking Steinert's disease with clinical
deterioration through 3 generations and offering appropriate genetic counseling.
Introduction
La maladie de Steinert ou DM1 est une dystrophie musculaire caractérisée par une hétérogénéité clinique à la fois inter et intra familiale et par une atteinte pluriviscérale ; musculaire, respiratoire, endocrinienne, oculaire… et surtout cardiaque qui peut conduire à la gravité de la maladie et mettre en jeu le pronostic vital (Brook et al. 1992).
Le gène DMPK responsable de la maladie de Steinert est localisé sur le chromosome 19 (19q13.3), il code pour une protéine kinase (Eriksson et al., 2001). La mutation responsable est une expansion de triplets CTG dans la région 3’ non codante du gène (Amack et al., 1999).
L’allèle normal comporte
entre 5 et 35 répétitions CTG. A partir de 50 répétitions CTG l’allèle devient
morbide. Cette mutation se transmet de façon instable avec augmentation de la
taille de la répétition d’une génération à l’autre ce qui explique le phénomène
d’anticipation, qui se traduit par l’aggravation à travers les générations
suivant le model : cataracte à la première génération, forme commune chez la
deuxième et une forme congénitale chez la troisième génération. (Laura et al., 2004; Tan et
al., 2005)
Ces trois formes peuvent coexister dans une même famille, sur trois générations successives. Les formes congénitales sont toujours de transmission maternelle, même si récemment quelques rares cas de transmission paternelle ont été décrits. (Tan et al., 2005)
La DM1 reste la plus fréquente des dystrophies à l’échelle mondiale (Bouhour et al., 2007) avec une prévalence générale de 1 cas pour 20.000. Au Maroc nous n’avons pas de travail fait sur cette pathologie.
Dans ce travail nous présentant le diagnostic moléculaire et le conseil génétique de la maladie de Steinert chez une famille marocaine avec phénomène d’anticipation.
Observation
Notre étude a concerné au début un enfant de 3 mois qui présente depuis la naissance une hypotonie généralisée avec une détresse respiratoire (polypnée, tirage et cornage …) et une fatigue intense lors de la succion-déglutition, ce tableau a nécessité des hospitalisations répétées de l’enfant en unité de soins intensifs. L’interrogatoire de la mère rapporte la notion d’une faible motilité fœtale au cours de la grossesse, toutefois la biométrie et le poids au cours de la grossesse et à la naissance étaient sans anomalie.
L’enquête familiale a montré l’existence chez la mère d’une myotonie avec lourdeur musculaire, une gêne respiratoire intermittente. Une décompensation a été remarquée lors de la grossesse et l’accouchement avec apparition, en post-partum, d’une claudication intermittente de périmètre de marche non déterminé mais qui s’est gravement réduit selon la mère. Son frère présente des douleurs musculaires instables mises dans le compte de son activité physique professionnelle et traitées symptomatiquement par des anti-inflammatoires et des antalgiques locaux puis généraux. Sa mère présente une cataracte bilatérales et sous antidiabétiques oraux depuis 12 ans pour un diabète non insulinodépendant.
Devant le tableau clinique de l’enfant et l’histoire familiale, un diagnostic de la maladie de Steinert a été évoqué. L’analyse moléculaire du gène DMPK par PCR (Figure 1) et Southern–blot a révélé la présence d’un nombre de CTG de 1500 chez l’enfant (IV1), 870 CTG chez la mère (III2) et son frère (III3) et 400 CTG chez la grand-mère (II2) et un de ses frères (II5) qui est jusque là asymptomatique. Alors que le chez les autres membres de la famille (III3, II2, II3, II5) nombre de CTG varie entre 5 et 17 sur les 2 allèles (Figure 2).
Devant ces résultats, un bilan complet, notamment cardiaque, a été réalisé afin d’éliminer toute atteinte qui engage le pronostic vital même dans les formes asymptomatiques.
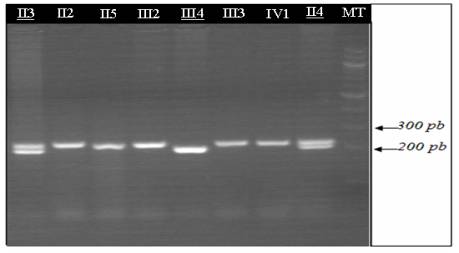
Figure 1. Gel d’agarose 3% qui montre le profil des membres de la
famille atteints de DM1.
MT : marquer de taille. Les Malades II2, II5, III2, III3, IV1
présentent une seule bande. Les
sujets normaux II3, II4 présentent 2 bandes (hétérozygotes). III4 présente 1
bande intense (homozygote).
Figure 1. 3% agarose gel which shows the profile of family members affected by DM1. MT: Weight Marker. Patients II2, II5, III2, III3, IV1 have a single band. Normal subjects II3, II4 have two bands (heterozygous). III4 presents a strong band (homozygous).
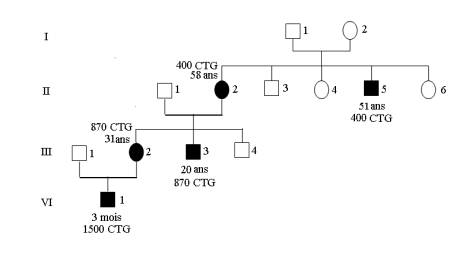
Figure 2. L’arbre généalogique de la famille atteinte de la maladie de Steinert
avec l’âge et le nombre de répétitions CTG.
Figure 2. Pedigree of the family with myotonic dystrophy , age and number of repetitions CTG.
Discussion
La maladie de Steinert est une affection de transmission autosomique dominante avec anticipation, un patient a donc une probabilité de 50% de transmettre la maladie à chacun de ses enfants indifféremment du sexe.
Cependant, deux phénomènes rendent le conseil génétique parfois délicat ; d’abord la grande variabilité phénotypique corrélée au nombre de répétitions de triplets CTG, et le phénomène d’anticipation qui est lié essentiellement au sexe du parent transmetteur de la mutation (Brunner et al., 1993).
Dans notre cas le nombre de répétions CTG est passé de 400 CTG à 870 CTG de la mère (II2) à la fille (III2) et de 870 CTG à 1500 CTG de la fille (III2) à son fils (IV1), ceci va dans le même sens d’une augmentation de la taille de la mutation dynamique dans le cas d’une transmission maternelle rapporté par G helen et al. 1993 . Pour le nourrisson (IV1), le passage d’une forme de l’adulte chez la mère à une forme néonatale plaide pour une autre augmentation du nombre de triplets d’autant plus que la forme néonatale est corrélée à un nombre de CTG supérieur à 1000 et que un nombre de CTG au-delà de 1500 défini la forme néonatale grave , létale dans 17 a 41 % par détresse respiratoire et associée dans 60% des cas à un retard mental (Longman, 2006).
Notre étude rejoint les conclusions de H. G. Brunner et al. 1993, concernant, le phénomène d’anticipation qui se traduit par une aggravation de profil clinique et une augmentation du nombre de CTG chez les malades à travers les générations, ce phénomène est lié à la transmission maternelle de la mutation qui détermine, elle seule, à un moment donné, une forme congénitale de pronostic réservé. D’autre coté, l’enquête familiale réalisée a permis de détecter un sujet porteur de la mutation et asymptomatique (II5) et qui peut développer à tout moment des symptômes, donc il bénéficie dorénavant d’une surveillance complète nettement cardiaque.
Conclusion
La prise en compte des 2 facteurs, l’anticipation en cas d’attente maternelle et surtout la corrélation entre le nombre de répétitions CTG et le profil clinique nous aide, lors du diagnostic prénatal (DPN), à répondre avec prudence à la question ‘qu’elle est le profil génétique et clinique du l’enfant à naître ?’ mais il reste toutefois très difficile de prédire avec précision la sévérité potentielle de la maladie.
Bibliographie
Amack J. D ; Paguio A. P.;
Mahamadvan M. S, 1999 Cis and trans effects of the myotonic dystrophy (DM)
mutation in a cell culture model, Human molecular genetics
ISSN 0964-6906 , vol. 8, no11, pp. 1975-1984
Bouhour F, Bost M, Vial C. Avril 2007 , Maladie de Steinert,Orphanet
Brook JD, McCurrach ME, Harley HG, et al. 1992, Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member., Cell. ; 68(4),799-808.
Brunner H. G., Brüggenwirth H. T,
Nillesen W. et al 1993, Influence of sex of the transmitting
parent as well as of parental allele site on the CTG expansion in myotonic
dystrophy (DM) , Am J Hum Genet. 53(5), 1016–1023.
Eriksson M.; Hedberg B.; Carey
N.; Ansved T. September 2001, Decreased DMPK Transcript Levels in Myotonic
Dystrophy 1 Type IIA Muscle Fibers,BBRC, Volume 286, Number 5, , pp.
1177-1182
Laura PWR, John WD. 2004; Pathogenic RNA repeats: an expanding role in genetic disease. Volume 20, Issue 10, Pages 506-512.
Longman
C, 2006, Department of Clinical Genetics, Guy’s Hospital, London, England,
Myotonic dystrophy, J R Coll
Physicians Edinb; 36,51–55
Tan EC; Lai, PS. 2005, Molecular diagnosis of neurogenetic disorders involving trinucleotide repeat expansions.Expert Review of Molecular Diagnostics; Vol. 5, No. 1, Pages 101-109 .
![]()